# FairChem
# 模块说明
基于 Meta FAIR Fairchem 框架、搭载原子通用模型(UMA)的高性能分子优化工具。
用户输入分子 3D 结构,工具即可在原子级精度下,快速计算能量、原子受力等关键物理量并自动完成结构优化。
# 使用步骤
# 上传输入文件
上传输入分子结构文件,支持xyz,sdf, mol2等格式。支持多个文件同时计算,每个文件包含一个分子3D结构。如果是一个文件包含多个分子结构,需要进行拆分后再进行上传。
# 设置模块运行参数
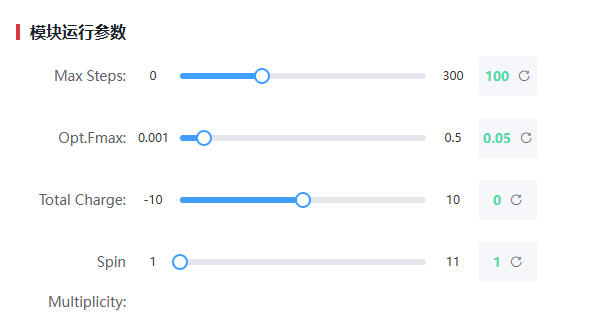
请通过拖动下方滑块来调整各项输入参数。每个滑块右侧显示当前设定值,最右侧绿色框内为默认或推荐值,点击刷新图标可快速恢复默认设置。
- Max Steps(最大步数):控制计算迭代的最大次数,范围 0–300,默认 100。
- Opt.Fmax(优化力阈值):决定结构优化的收敛标准,范围 0.001–0.5 eV/Å,默认 0.05。
- Total Charge(总电荷):设置体系净电荷,范围 -10 至 +10,默认 0。
- Spin Multiplicity(自旋多重度):指定自旋多重度,范围 1–11,默认 1。
# 提交计算
点击提交按钮,提交计算
# 结果下载
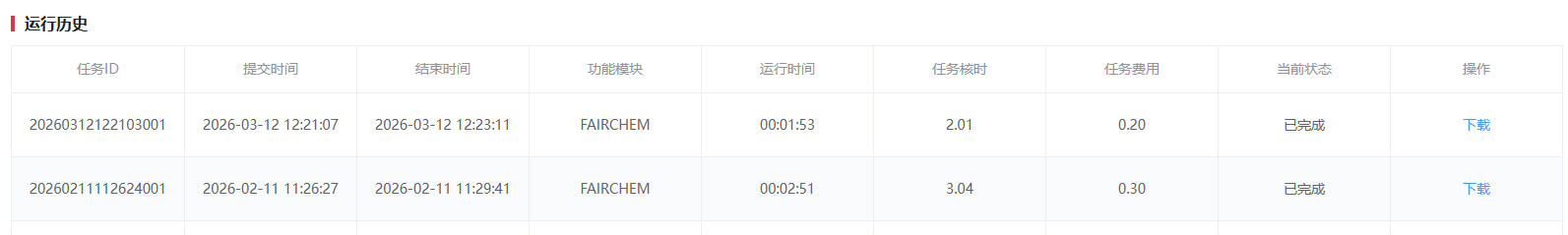
- 等待计算任务运行完成。当【当前状态】列显示为“已完成”时,表示任务已成功结束。
- 点击对应任务行右侧的【下载】按钮,即可获取计算结果文件。
- 若页面未及时更新状态,请手动刷新浏览器页面(快捷键 F5 或 Ctrl+R / Cmd+R),以同步最新任务进度。
# 结果展示
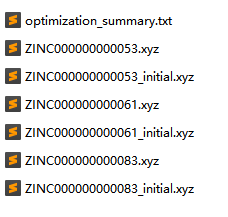
解压 result.tar 后,您将在 output 目录中看到以下关键文件:
🔹optimization_summary.txt → 包含全局计算参数、各分子优化能量与耗时明细、以及整体任务成功率的综合摘要报告。
🔹 .xyz 结构文件
- 文件名格式:xxxxxxxxxx.xyz→ 对应优化完成后的分子结构
- 文件名格式:xxxxxxxxxx_initial.xyz → 对应输入的初始分子结构