# 分子对接
#
# 概述
分子对接模块通过计算候选分子在靶蛋白结合位点中的最佳构象和亲和力,筛选出可能具有高结合潜力的药物分子。该模块对靶蛋白和配体分子进行几何计算,识别配体在蛋白结合口袋中的“结合姿势”,并基于打分进行筛选排序。这种虚拟筛选方法在药物研发的前期筛选中,能够有效节约时间和成本。
# 使用步骤
打开神农量子云平台 (opens new window)【虚拟筛选-分子对接】模块。
# 步骤1:输入蛋白质结构文件
输入PDB ID:可直接输入靶蛋白的PDB ID(例如:RCSB 6LU7 (opens new window))。
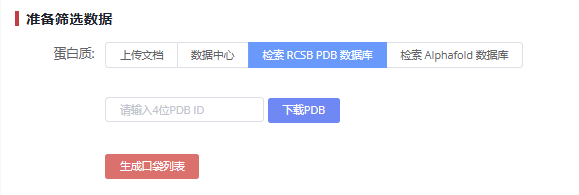
上传.pdb格式文件:如用户有自定义的蛋白质结构数据,上传蛋白质文件。

格式要求:标准PDB格式。
建议预处理:去除水分子、添加氢原子。 使用【靶点预处理】模块。
选择结合口袋:用户可以根据蛋白质的结构信息,指定结合口袋的位置。

系统会自动识别潜在的活性位点。
注意事项:优先选择打分靠前的口袋,准确的定义口袋可显著提高对接效率。
# 步骤2:输入配体文件
- 上传配体文件
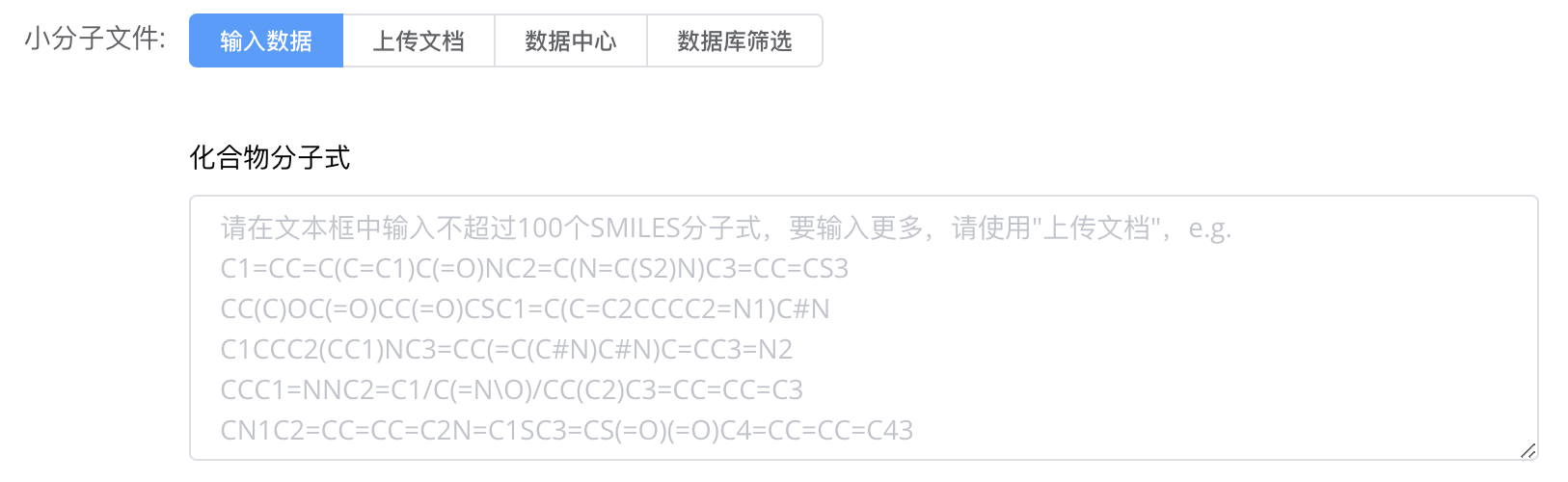
1.1 输入SMILES
文件中每行包含一个小分子的SMILES格式结构。以下是SMILES示例:
O=C(O[C@@H]1C[C@@H]2C[C@H]3C[C@H](C1)N2CC3=O)c1c[nH]c2ccccc12
CN/C(=C\[N+](=O)[O-])NCCSCc1ccc(CN(C)C)o1
C#C[C@]1(OC(C)=O)CC[C@H]2[C@@H]3CCC4=C[C@@H](OC(C)=O)CC[C@@H]4[C@H]3CC[C@@]21C
CCCCC1=NC2(CCCC2)C(=O)N1Cc1ccc(-c2ccccc2-c2nn[nH]n2)cc1
CC(C)(C)c1ccc(cc1)C(=O)Nc1cccc(c1)c1ccc(cc1)O
Cc1ccc(cc1)C(=O)Nc1c2ccccc2nc1
1.2 上传CSV文件
配体文件以.csv格式上传,表头为ID,SMILES。以下是CSV实例:
ID,SMILES
CHEM_01,O=C(O[C@@H]1C[C@@H]2C[C@H]3C[C@H](C1)N2CC3=O)c1c[nH]c2ccccc12
CHEM_02,CN/C(=C\[N+](=O)[O-])NCCSCc1ccc(CN(C)C)o1
CHEM_03,C#C[C@]1(OC(C)=O)CC[C@H]2[C@@H]3CCC4=C[C@@H](OC(C)=O)CC[C@@H]4[C@H]3CC[C@@]21C
CHEM_04,CCCCC1=NC2(CCCC2)C(=O)N1Cc1ccc(-c2ccccc2-c2nn[nH]n2)cc1
CHEM_05,CC(C)(C)c1ccc(cc1)C(=O)Nc1cccc(c1)c1ccc(cc1)O
CHEM_06,Cc1ccc(cc1)C(=O)Nc1c2ccccc2nc1
# 步骤3:设置对接参数
对接精度:指定搜索算法的精细度。更高的精度值会提高搜索深度,通常在5到32之间调整,以平衡速度与准确性。
结合姿势数:生成的配位模式数量,表示每个配体与靶标结合的不同构象数。更多的构象搜索会增加计算时间。
筛选个数:根据结合能筛选出的最佳构象数,并保存为pdb格式文件,便于进一步分析和用于结合自由能预测模块。
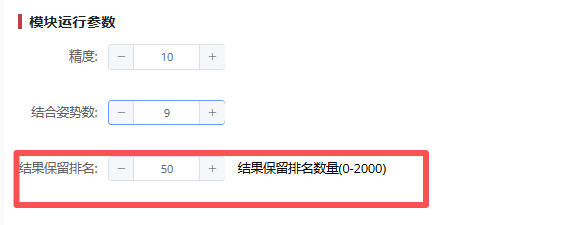
提交任务:完成参数设定后,点击【提交】按钮开始计算。
# 结果说明
文件说明:计算结果生成一个包含对接信息的文件夹压缩tar包,主要内容如下:
scores.json:包含每个配体的对接打分。ligands/:口袋文件夹下包含筛选对接分数高的配体的对接构型文件(.mol2格式),压缩为zip格式存储。protein/:包含输入的蛋白质结构文件。pocket.pdb:口袋文件ligands.csv:配体分子筛选结果csv文件
# tar 包 output/ ├── scores.json ├── ligands.csv ├── pocket2_atm.pdb ├── pocket3_atm.pdb ├── ligands/ │ └── ligands_mol2.zip └── protein/ └── 6lu7.pdb # 配体三维结构zip 压缩包 ligands_mol2.zip ├──pocket2_atm/ # 口袋同名文件夹 │ ├── 644196.mol2 │ ├── 53361968.mol2 │ └── 451415.mol2 └── pocket3_atm/ # 口袋同名文件夹 ├── 15942730.mol2 ├── 441243.mol2 ├── 94736851.mol2 └── 134815261.mol2结果介绍:
scores.json文件记录了各配体的打分数据,结合分值越低表示结合更稳定。
参数名称 参数介绍 备注 ID 分子标识 计算中唯一,来源多为分子库标识,通过从配体csv文件中读取 SMILES 分子式 通过从配体csv文件中读取,需要使用标准SMILES格式 score 对接分数 单位kcal/mol,用于估算亲和力。集合形式,通过对接参数获取的对接结果分数,集合对应配体姿势打分 pocket 对接口袋名称 输入或选择的口袋名称 part 并行对接的区块 可不关注 使用引导 根据筛选的结果,可以进一步筛选并优化候选分子,可以使用平台模块【结合自由能】【药物-靶点相互作用3D】继续药物开发的后续筛选计算。
← 药效团 药物-靶点相互作用(3D) →